



About
PEPSI-Dock (Polynomial Expansion of Protein Structures and Interactions for Docking) is a protein-protein docking prediction algorithm that outputs a list of most-possible conformations, taking as inputs two known structures. It relies on the Hex exhaustive search1 and a very detailed data-driven model of the binding free energy2 under the assumption of rigid bodies and linear interactions of the atoms composing the interface. It is used at the very first stage of the docking pipeline and will be avalaible soon as a SAMSON Module.
Method
PEPSI-Dock combines an exhaustive search algorithm that finds the possible complexes formed by two macromolecules with a detailed data driven protein-protein interaction potential to compute the binding free energy of these complexes. The exhaustive search algorithm is provided by the Hex library, which uses FFT-accelerated techniques in the Spherical Fourier Polar basis.
The accuracy of the algorithm is achieved thanks to a set of knowledge-based interaction potentials.
We should mention that the computation of the interaction potentials is performed only once off-line, during the learning phase. Then these are stored and loaded with no cost when doing the search.
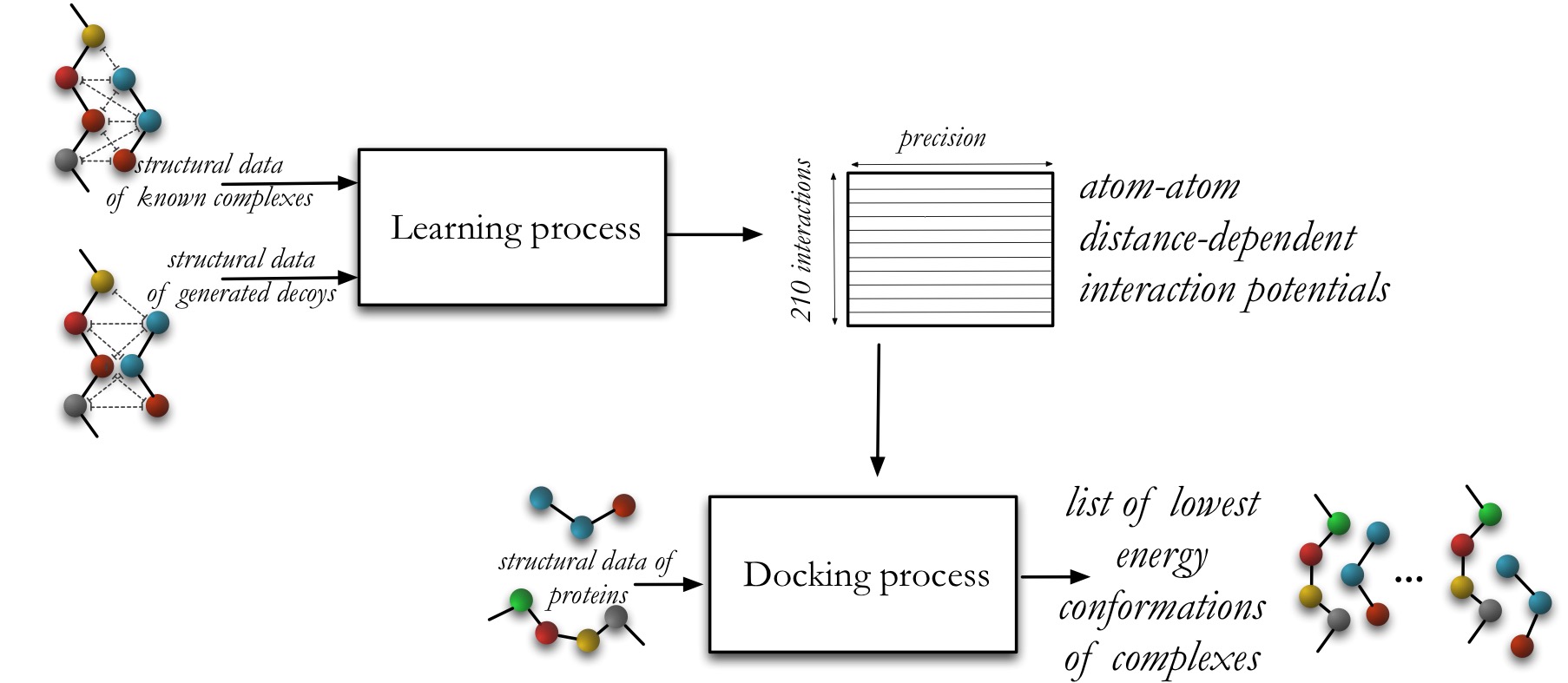
Illustration of the main concepts of the PEPSI-Dock method, which consists in two stages.
The first stage is the learning phase, where starting from a set of native and non-native complexes, we deduce 210 atomic 1-D distance-dependent interaction potentials.
These potentials are based on commonly-used physical assumptions accounting for 210 different types of interactions that depend on the atom characteristics (chemical element, aromaticity, polarity, etc).
They are constructed with the specific objective of discriminating native structures from the non-native ones, thanks to the use of a rigorous learning optimisation algorithm inspired by the Support Vector Machine (SVM) method.
The feature space for the optimization problem consists in geometry and the chemistry features extracted from interfaces between proteins.
The second stage is the docking prediction phase, where starting from a pair of macromolecules, a list of putative binding poses corresponding to the lowest binding free energy conformations is computed.
The possible conformations are explored using a global exhaustive search algorithm. It represents the protein geometry through 20 3-D Gauss-Laguerre expansion coefficients and uses the Spherical Polar Fourier transform to rapidly compute the energy overlap integrals.
Data sets used in this work
The training set is defined as the set of 195 heterodimers from the 851 non-redundant protein-protein complex structures prepared by Huang and Zou Huang and Zou (2008). This database contains protein-protein complexes extracted from the PDB Berman et al. (2000) and includes 655 homodimers and 195 heterodimers.
For the test set we used the Protein Docking Benchmark, which is the most widely used set of protein complexes collected both in the bound and in the unbound forms. Version 5.0 of the benchmark was released recently (Vreven et al., 2015) and consists of 230 protein complexes.
Results
We compared PEPSI-Dock performance with the original method implemented in Hex with the shape complementarity plus electrostatics scoring function, starting from bound complexes on the training set of 162 hetero-dimers. PEPSI-Dock achieves the success rate of 91% mid-quality predictions found in top-10 solutions whereas original Hex reaches 43,1 %.
On a subset from the protein docking benchmark v5, it achieves 73% mid-quality predictions found in top-10 solutions starting from bound structures or 19% if started from the unbound structures. Even though we did not train the potential to predict near-natives from unbound conformations, our method favourably competes with Hex and ZDOCK, even if ZDOCK has a finer sampling precision and uses a Cartesian sampling grid.
Running time
The method ranks about 109 putative binding poses in 5-10 minutes on a modern laptop for a mid-size protein.
Authors
Emilie Neveu, David W. Ritchie & Sergei Grudinin.
Nano-D team, Inria/CNRS Grenoble, France e-mail: Sergei.Grudinin @ inria.frDownload
PEPSI-Dock will be made available at https://www.samson-connect.net
License
All rights reserved. The academic version is free.
References
1 Accelerating and focusing protein–protein docking correlations using multi-dimensional rotational FFT generating functions, Ritchie, David W. and Kozakov, Dima and Vajda, Sandor, Bioinformatics, 24(17), 1865-1873, 2008
2 PEPSI-Dock : A Detailed Data-Driven Protein-Protein Interaction Potential Accelerated By Polar Fourier Correlation, Neveu E., Ritchie, David W., Popov P., and Grudinin S. Bioinformatics, Oxford University Press (OUP), 2016, <10.1093/bioinformatics/btw443>.