



About
NOn-Linear rigid Block NMA approach (NOLB) a new conceptually simple and computationally efficient method for non-linear normal mode analysis.

Method
The key observation of our method is that the angular velocity of a residue can be interpreted as the result of an implicit force, such that the motion of the residue can be considered as a pure rotation about a certain center.
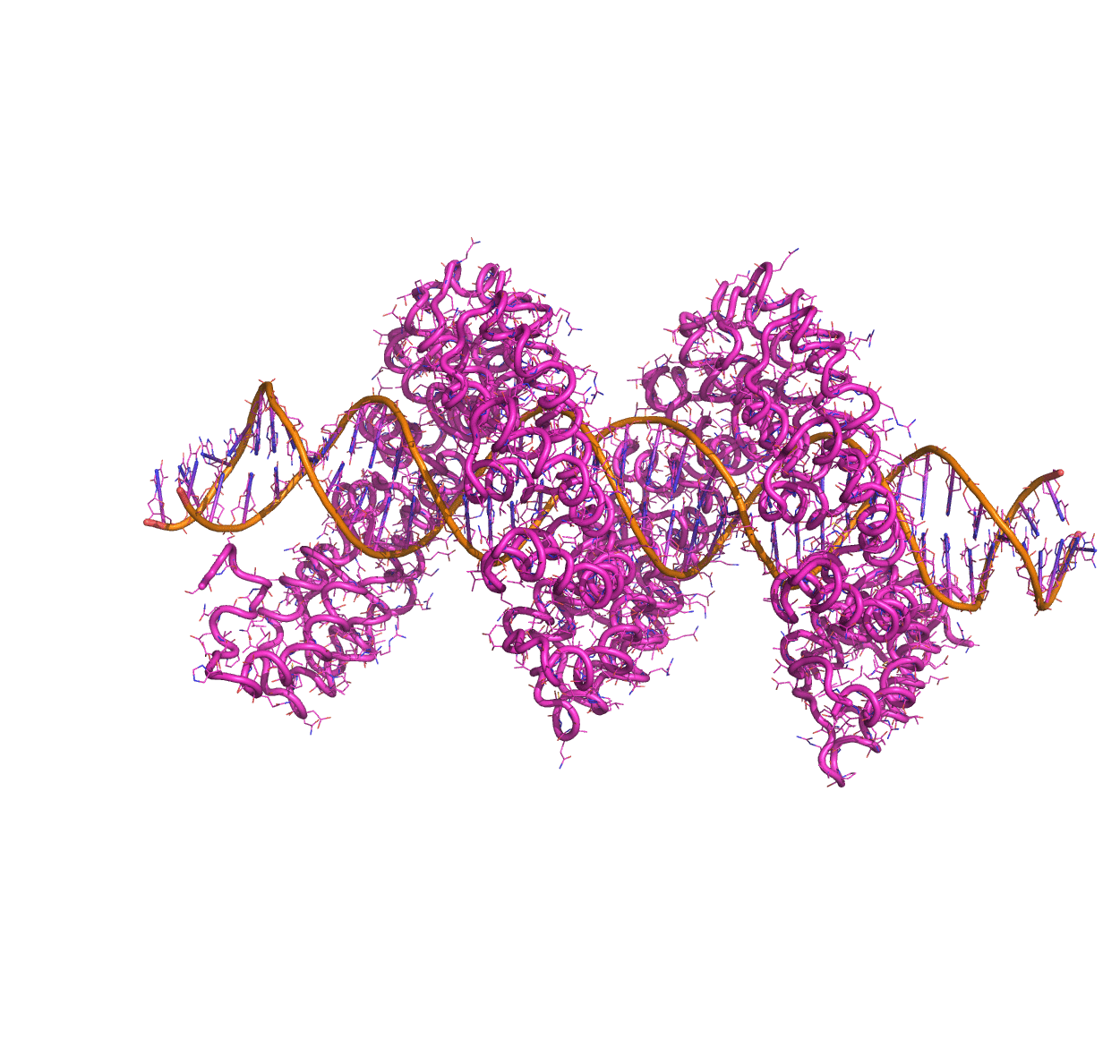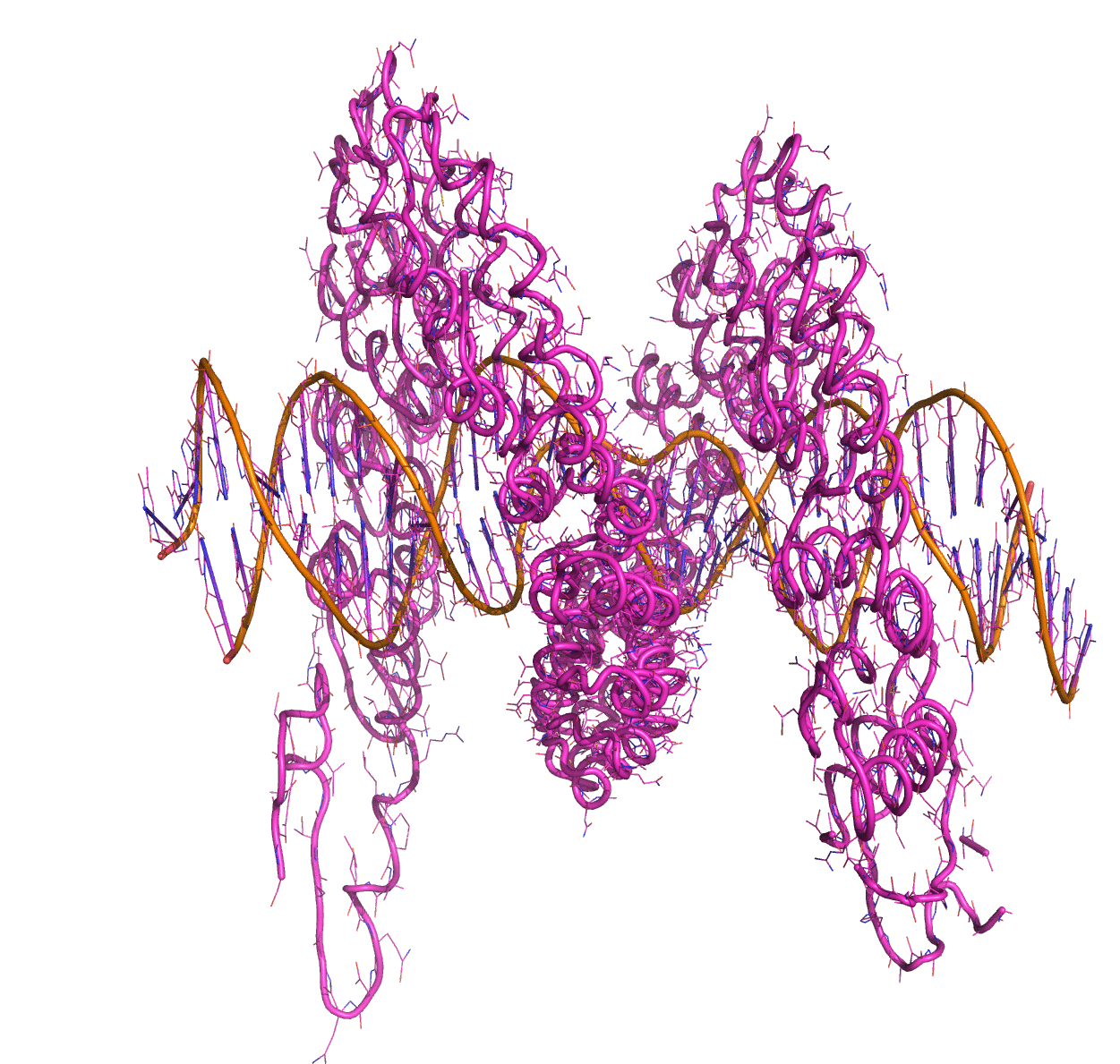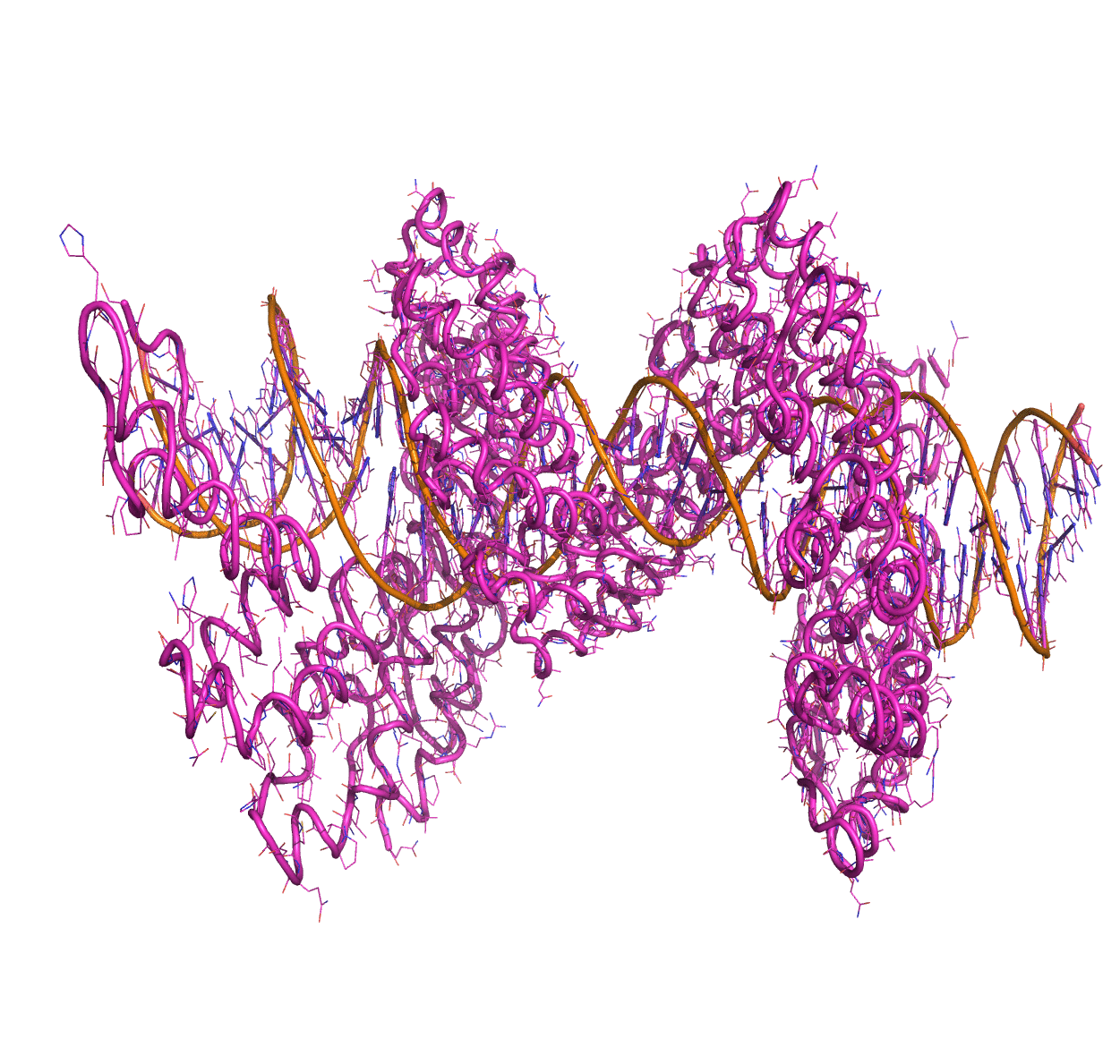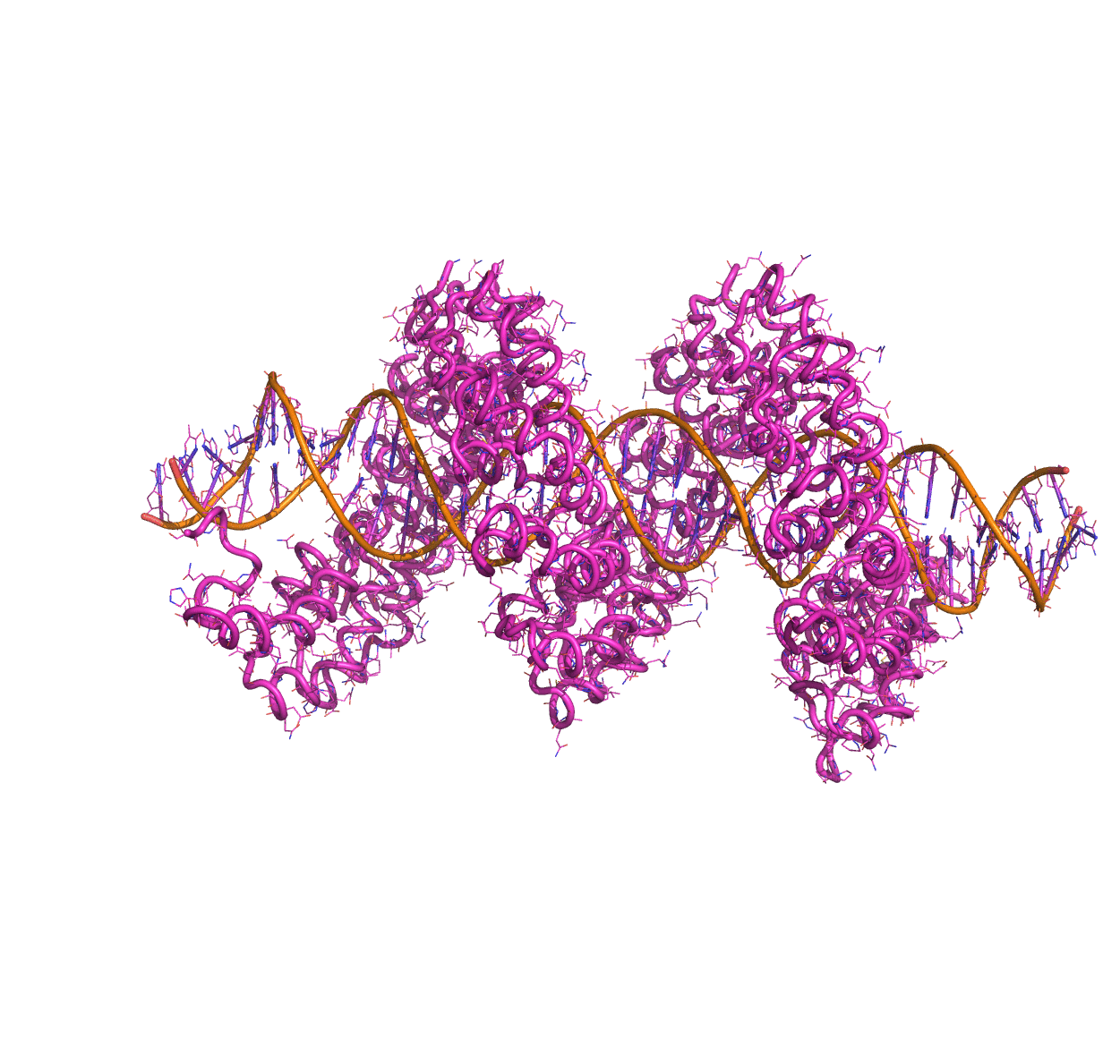 Linear Normal Modes of a protein-DNA complex |
 Non-Linear Normal Modes of a protein-DNA complex |
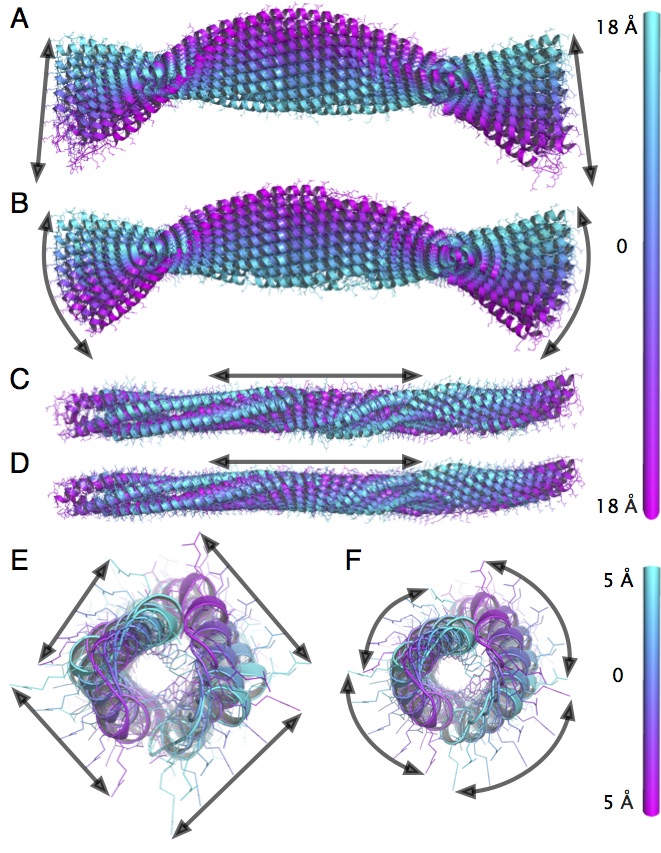

Authors
Alexandre Hoffmann & Sergei Grudinin,
Nano-D team, Inria/CNRS Grenoble, France e-mail: Sergei.Grudinin @ inria.frDownload
NOLB User Guide (v. 1.1)
NOLB for MacOS (v. 1.9)
NOLB for Linux (v. 1.9, supported GLIBC versions >= 2.10)
Data sets and scripts for structural transitions
SAMSON GUI
GUI will be made available soon at https://www.samson-connect.net
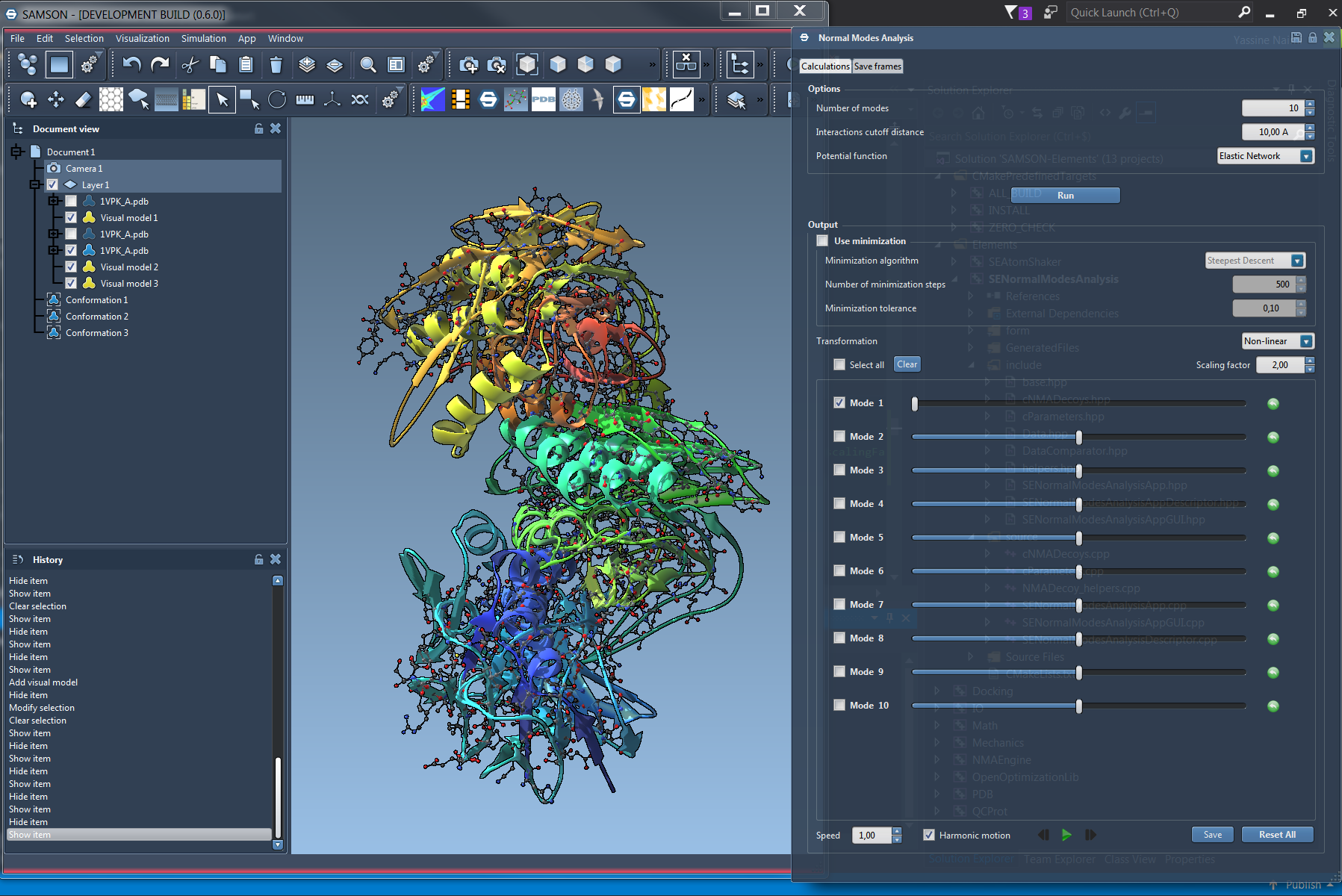
SAMSON NOLB element
Usage cases and examples
For the explanation of the main method please see [1]. The way to generate structural ensembles within constant RMSD and applications to flexible docking are shown in [2]. Nonlinear structural transitions including those with iterative reconstruction of the Hessian matrix are explained and assessed in [3]. Elastic network modification using coloured contact maps, called HOPMA (Cyrillic transcript for NORMA), which boosts functional structural transitions and dynamics is given in [4]. We plan to integrate the essence of the HOPMA code inside NOLB, the full package is available at https://github.com/elolaine/HOPMA.
License
All rights reserved. The academic version is free.
References
[1] Alexandre Hoffmann & Sergei Grudinin. NOLB : Non-linear rigid block normal mode analysis method. Journal of Chemical Theory and Computation, 2017, 13 (5), pp.2123-2134. DOI: 10.1021/acs.jctc.7b00197. 

[2] Emelie Neveu, Petr Popov, Alexandre Hoffmann, Angelo Migliosi, Xavier Besseron, Gregoire Danoy, Pascal Bouvry, Sergei Grudinin. RapidRMSD: Rapid determination of RMSDs corresponding to motions of flexible molecules. Bioinformatics, Oxford University Press (OUP), 2018, 34(16):2757–2765.〈10.1093/bioinformatics/bty160〉
[3] Sergei Grudinin, Elodie Laine & Alexandre Hoffmann. Predicting protein functional motions: an old recipe with a new twist. 2020. Biophysical Journal. Volume 118, Issue 10, 19 May 2020, Pages 2513-2525. https://doi.org/10.1016/j.bpj.2020.03.020.
[4] Elodie Laine & Sergei Grudinin. HOPMA: Boosting protein functional dynamics with colored contact maps. 2020. https://doi.org/10.1101/2020.12.31.424963.
Other NMA approaches
There is a list of other NMA methods including the following,
- The original RTB method by Y.H. Sanejouand and its applications at http://www.sciences.univ-nantes.fr/elnemo/modes.html
- IMOD NMA in internal coordinates by Pablo Chacón at http://chaconlab.org/multiscale-simulations/imod
- NMA applications by Marc Delarue at http://lorentz.dynstr.pasteur.fr/nomad-ref.php
- ProDy Python package for protein structural dynamics analysis by Ivet Bahar at http://prody.csb.pitt.edu