



About
KNOwledge-Driven Ligand Extractor is a software library for the recognition of atomic types, their hybridization states and bond orders in the structures of small molecules. Its prediction model is based on nonlinear Support Vector Machines.
Method
The process of bond and atom properties perception is divided into several steps. At the beginning, only information about the coordinates and elements for each atom is available :
- Connectivity is recognized.
- A search of rings is performed to find the Smallest Set of Smallest Rings (SSSR).
- Atomic hybridizations are predicted by the corresponding SVM model.
- Bond orders are predicted by the corresponding SVM model.
- Aromatic cycles are found.
- Atomic types are set in obedience to the functional groups. Some bonds are reassigned during this stage.
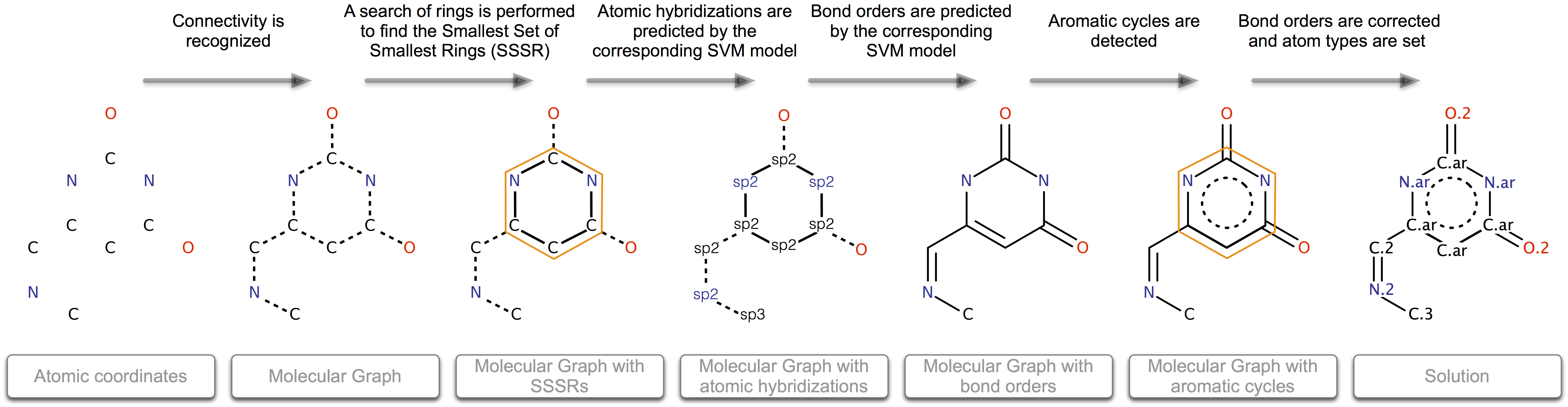
Figure 1: Schematic workflow of the Knodle algorithm.
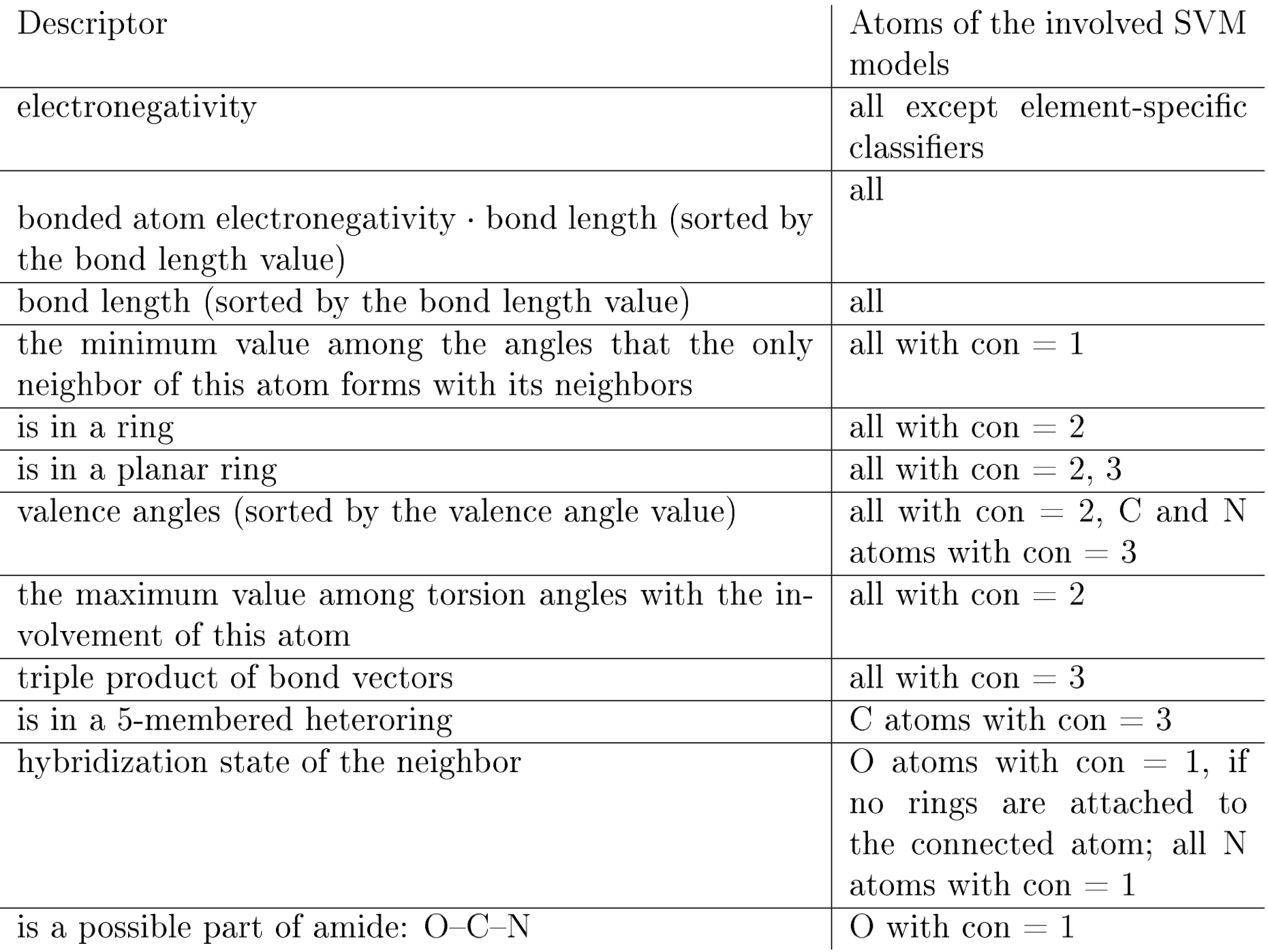
Table 1: Descriptors for the prediction of hybridization states. Each descriptor is applicable to one or more atom classification groups. An atom is assigned to a classification group according to the number of its heavy neighbors and its chemical element. Here, “con” stands for the “connectivity” parameter of an atom — the number of its heavy neighbors.

Table 2: Descriptors for the prediction of bond order. Indices 1 and 2 correspond to the first and the second atoms involved in the bond.
Table 3 lists the classifiers and the results of their training.
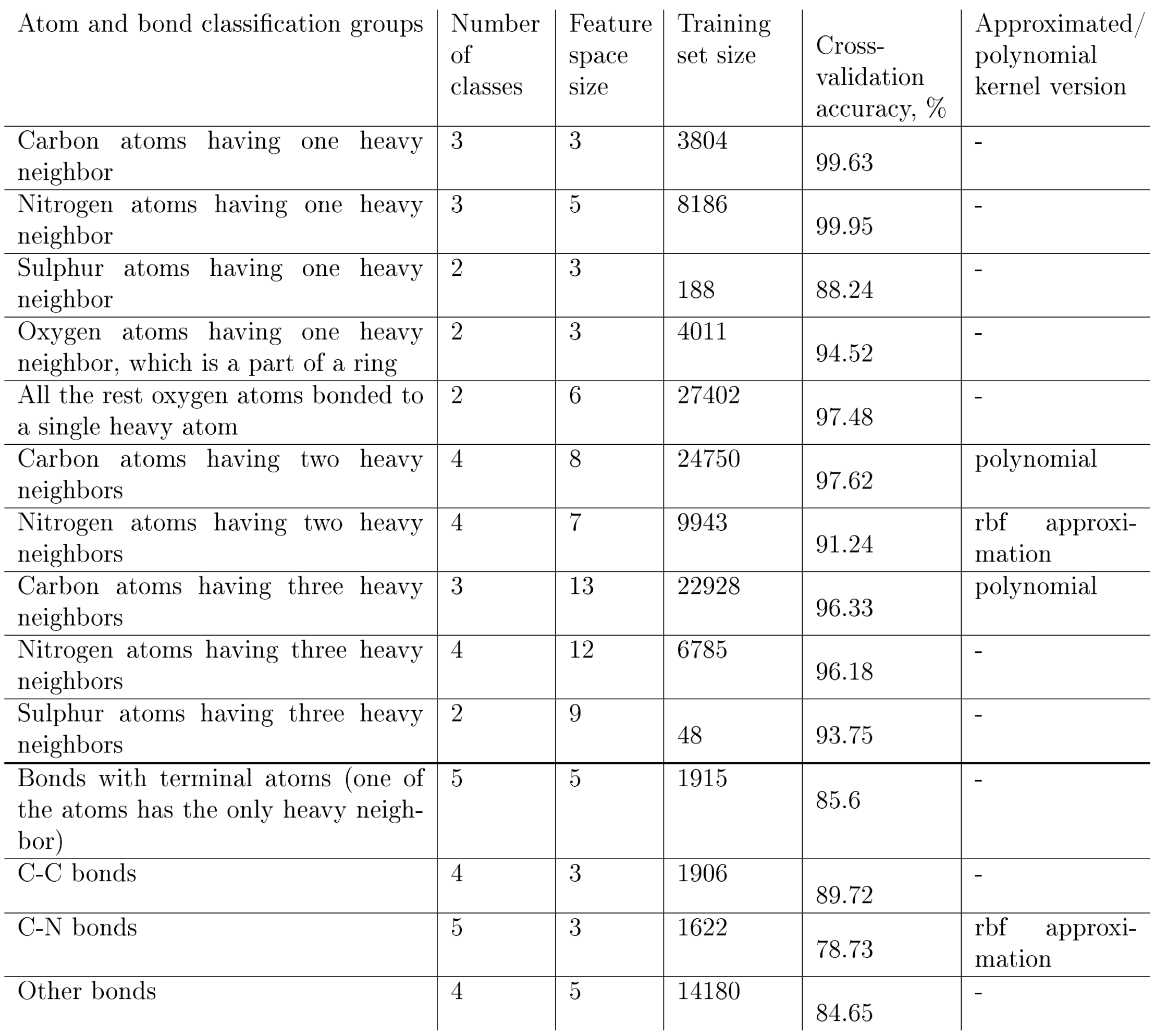
Table 3: Classification groups for atoms and bonds that have the corresponding SVM models, number of classes, feature space size, the training set size, the training results, and the SVM kernels used in our model. ‘Rbf approximation’ stands for the second-order Maclaurin series approximation of the radial basis kernel. ‘Polynomial’ stands for the third-order polynomial approximation of the radial basis kernel. The ‘Cross-validation accuracy’ column gives the ratio of correctly predicted hybridizations and bond orders measured on the training set after a 5-fold cross validation.
Test cases
We compared Knodle’s performance with three popular methods for molecular types recognition and format conversion, NAOMI, fconv, and I-interpret. To do so, we ran several series of tests. First, we assessed the accuracy of the methods on the Labute’s benchmark set of 179 protein-ligand complexes. Second, we assessed the accuracy of the methods on a part of the PDBBindCN general data set (v2014) consisting of 3,000 protein- ligand complexes that were randomly excluded from the SVM model training. Third, we performed a more general evaluation of our method using 332,974 ligands extracted from the Ligand Expo database. Finally, we compared the running times of the methods using 10,605 protein-ligand complexes from the PDBBindCN general data set (v2014), and 10,605 isolated ligands from the same dataset.
Labute’s test set
Overall, on the Labute’s benchmark set, Knodle made five to six perception errors depending on the chosen SVM kernels, NAOMI made seven errors, fconv made thirteen errors, and I-interpret made nine errors.
PDBBindCN test set
Overall, fconv demonstrated the worst performance with approximately 12.8% of errors. Knodle performs slightly better than NAOMI having 3.9% versus 4.7% of errors, correspondingly, while I-interpret made 6.0% of errors.
Ligand Expo Test Set
Overall, 15,028 entries out of 332,974 are perceived incorrectly, accounting for 4.5% of the initial dataset.
Running time
We performed a comparison of running times of the Knodle, fconv, NAOMI and I-interpret methods using the PDBBindCN general data set (v2014) consisting of 10,605 full protein-ligand complexes and 10,605 extracted ligands. The tests were run on the following machines, a desktop with Linux and 3.10 GHz Intel(R) Core(TM) i5-4440 CPU processor and 16 GB 1600 MHz DDR3 RAM, and a MacBook Pro late 2013 laptop with a 2.6 GHz Intel Core i7 processor and 16 GB 1600 MHz DDR3 RAM. Table 4 lists the timings measured on two platforms. Overall, we can see that when reading large files, Knodle is significantly faster than the other methods. However, when converting small files, all the four tested methods demonstrate a very similar performance with fconv being the fastest one, which is however negated by its accuracy.
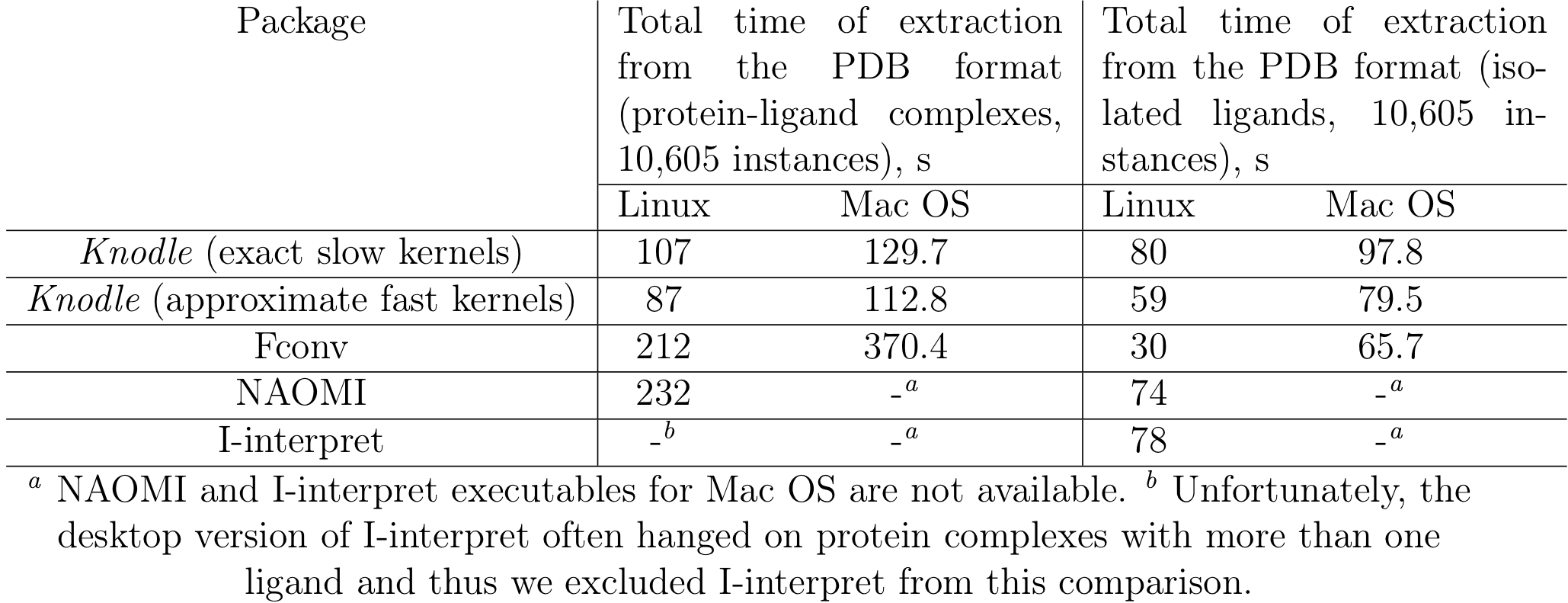
Table 8: Running times of the Knodle, fconv, NAOMI, and I-interpret methods for the PDBBind 2014 dataset. Timings for two series of tests are given, the first for 10,605 protein- ligand complexes, and the second for 10,605 isolated ligands. Running times for Knodle are given for the two versions of its SVM kernels, the exact one and the approximate one.
Data sets used in this work
A part of the PDBBindCN general data set (v2014) consisting of 3,000 protein- ligand complexes that were randomly excluded from the SVM model training Errors of the tested methods on the PDBBindCN dataset Labute’s benchmark set of 179 protein-ligand complexes 10,605 isolated ligands from the PDBBindCN general data set (v2014) Ligand Expo test set of 332,974 ligands 25 most frequent ligands from the LigandExpo dataset in which Knodle conversion errors occurred
Authors
Maria Kadukova1 & Sergei Grudinin2,
1Laboratoty of Advanced Studies of Membrane Proteins, MIPT Moscow, & 2Nano-D team, Inria/CNRS Grenoble, France e-mail: Sergei.Grudinin @ inria.fr
Download (version 0.9)
Knodle for Linux (v. 0.9)
Download (version 0.3)
Knodle for MacOS (v. 0.3)
Knodle for Linux (v. 0.3)
Knodle for Windows is coming soon
Knodle GUI will be available at https://www.samson-connect.net
License
All rights reserved. The academic version is free.
Reference
Maria Kadukova & Sergei Grudinin, “Knodle, a Support Vector Machines-based automatic perception of organic molecules from 3D coordinates”, J. Chem. Inf. Model., 2016, DOI: 10.1021/acs.jcim.5b00512.