
Description
Compareads is the first version of a fast software for comparing two sets of metagenomics data. The output is a score of similarity.
Each set may include hundreds of millions reads.
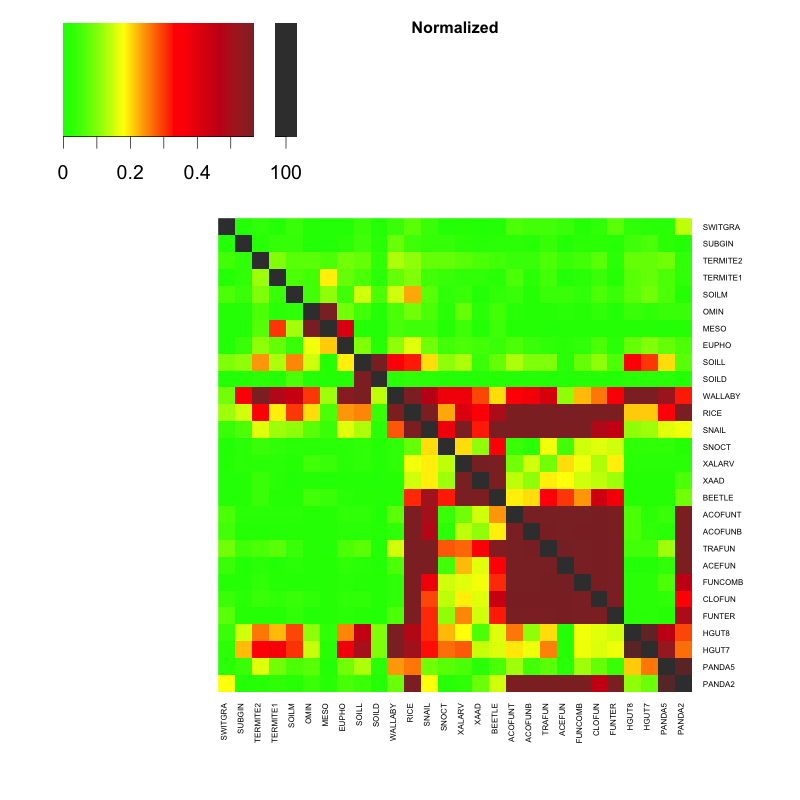
Commet is the next version allowing multiple sets of metagenomic to be combined together. The output is a matrix of scores.
Web Site
Reference
N. Maillet, G. Collet, T. Vannier, D. Lavenier, P. Peterlongo, COMMET: comparing and combining multiple metagenomic datasets, IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2014.
abstract Metagenomics offers a way to analyze biotopes at the genomic level and to reach functional and taxonomical conclusions. The bio-analyzes of large metagenomic projects face critical limitations: complex metagenomes cannot be assembled and the taxonomical or functional annotations are much smaller than the real biological diversity. This motivated the development of de novo metagenomic read comparison approaches to extract information contained in metagenomic datasets. However, these new approaches do not scale up large metagenomic projects, or generate an important number of large intermediate and result files. We introduce COMMET (“COmpare Multiple METagenomes”), a method that provides similarity overview between all datasets of large metagenomic projects. Directly from non-assembled reads, all against all comparisons are performed through an efficient indexing strategy. Then, results are stored as bit vectors, a compressed representation of read files, that can be used to further combine read subsets by common logical operations. Finally, COMMET computes a clusterization of metagenomic datasets, which is visualized by dendrogram and heatmaps.
N. Maillet, C. Lemaitre, R. Chikhi, D. Lavenier, P. Peterlongo, Compareads: comparing huge metagenomic experiments. RECOMB Comparative Genomics 2012, Oct 2012, Niterói, Brazil
abstract: Nowadays, metagenomic sample analyses are mainly achieved by comparing them with a priori knowledge stored in data banks. Even if powerful, such approaches do not allow to exploit unknown and/or unculturable species, for instance estimated at 99% for Bacteria. This work introduces Compareads, a de novo comparative metagenomic approach that returns the reads that are similar between two possibly metagenomic datasets generated by High Throughput Sequencers. One originality of this work consists in its ability to deal with huge datasets. The second main contribution presented in this paper is the design of a probabilistic data structure based on Bloom lters enabling to index millions of reads with a limited memory footprint and a controlled error rate. We show that Compareads enables to retrieve biological information while being able to scale to huge datasets. Its time and memory features make Compareads usable on read sets each composed of more than 100 million Illumina reads in a few hours and consuming 4Gb of memory, and thus usable on today’s personal computers.
 Inria
Inria